Gaucher disease overview
Last updated Jan. 21, 2026, by Marisa Wexler, MS
 Fact-checked by Inês Martins, PhD
Fact-checked by Inês Martins, PhD
Gaucher disease is a rare genetic disorder that can affect multiple parts of the body, causing issues such as enlarged organs, blood disorders, bone abnormalities, and, in some cases, neurological complications. There are three main types of Gaucher disease, distinguished based on the presence and severity of neurological issues.
While Gaucher disease cannot be cured, treatments are available to help manage symptoms in most cases. These therapies can extend life expectancy and improve quality of life for many people living with the condition.
What is Gaucher disease?
Gaucher disease is caused by mutations in the GBA1 gene, which lead to the accumulation of certain fatty molecules that drive organ damage and disease symptoms.
Gaucher disease is a rare disorder. The global Gaucher disease prevalence is estimated to be about 1.5 out of every 100,000 live births. The condition can affect people of all demographic backgrounds, but it’s particularly common in those of Ashkenazi Jewish descent, where about 1 out of 450 live births are affected.
Everyone inherits two copies of the GBA1 gene, one from each parent. Gaucher disease follows an autosomal recessive inheritance pattern, which means the disease will only develop if a person inherits two mutated copies of the gene.
People with one mutated copy and one healthy copy are known as Gaucher disease carriers because they will not develop the disease. But carriers may pass the mutated gene on to their biological children. If:
- two carriers reproduce, there is a 25% chance their child will have Gaucher disease, a 50% chance the child will be a carrier, and a 25% chance the child will have neither Gaucher disease nor be a carrier
- a Gaucher disease carrier reproduces with someone who has Gaucher disease, there’s a 50% chance their child will have Gaucher disease and a 50% chance the child will be a carrier.
Types of Gaucher disease
The three main Gaucher disease types are all caused by GBA1 mutations. What distinguishes them is the presence and severity of neurological symptoms. In Gaucher disease:
- Gaucher disease type 1 is marked by no neurological symptoms
- Gaucher disease type 2 is marked by severe, early-onset neurological problems that progress rapidly
- Gaucher disease type 3 features milder neurological issues that appear in childhood and progress more slowly.
Gaucher disease type 1 is by far the most common form of the disease, accounting for at least 90% of all cases. Most people start having symptoms in childhood, but about a third of patients won’t see any symptoms before adulthood. Available treatments can help manage most symptoms and, with appropriate care, life expectancy will be close to the general population.
Gaucher disease type 2 is much rarer, accounting for about 5% of cases. It’s characterized by severe and rapidly worsening neurological problems that appear within the first months after birth. There are no effective treatments for it, and most children don’t survive beyond age 2.
Gaucher disease type 3 is another rare form that represents about 5% of all Gaucher disease cases. It’s also marked by neurological symptoms, but these are milder, worsen more slowly, and may not develop until later in childhood. While existing treatments can help to control non-neurological disease manifestations in these patients, there are no treatment options for managing their neurological problems. For that reason, people with Gaucher disease type 3 may survive into adulthood wIth appropriate treatment, but their life expectancy is markedly reduced compared with the general population.
Causes
Gaucher disease is caused by GBA1 mutations that are present from the moment of conception. Therefore, the only true risk factor for developing it is having parents who have Gaucher or are carriers.
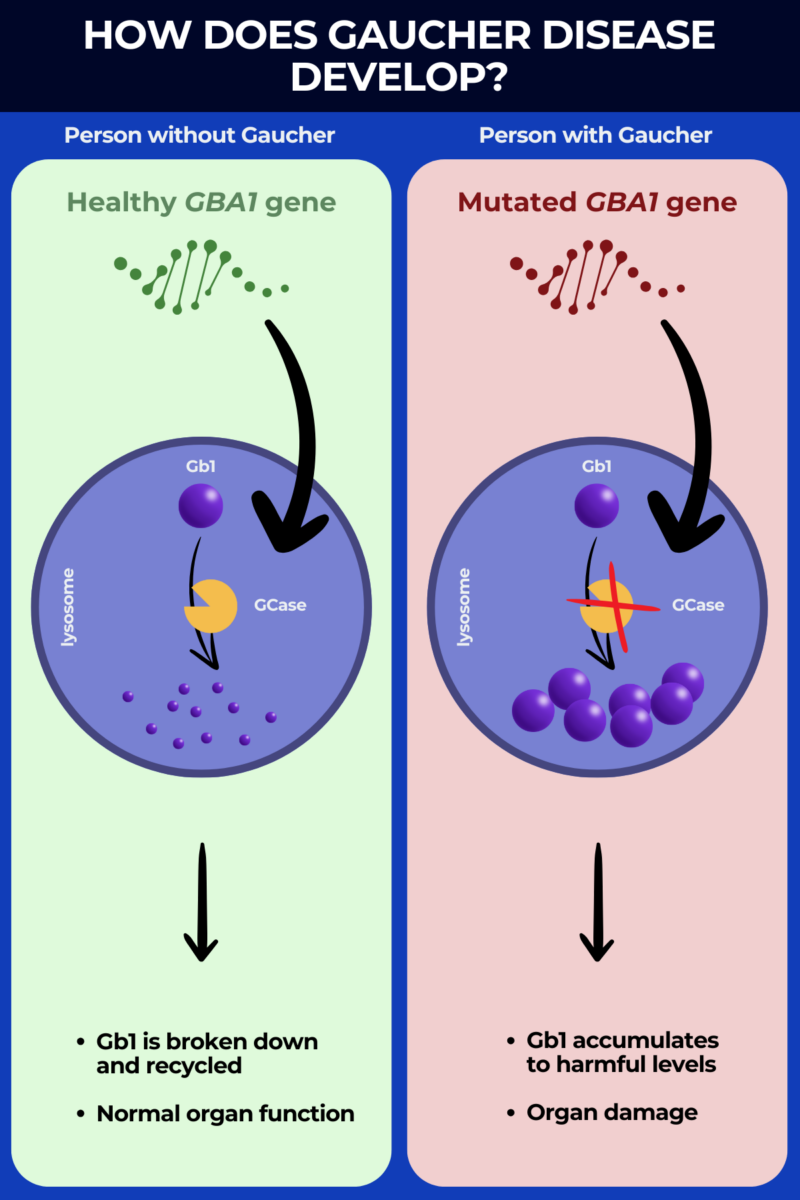
More than 400 mutations have been identified as Gaucher disease causes. These mutations interfere with the cells’ ability to produce the glucocerebrosidase (GCase) enzyme, which is needed to degrade a fatty molecule called glucocerebroside (Gb1).
When GCase is missing or not working properly, it cannot effectively break down Gb1, so this molecule accumulates to toxic levels inside certain cells. Gaucher disease enzyme deficiency particularly affects immune cells called macrophages, which are referred to as Gaucher cells when they become full of undigested Gb1.
These abnormal cells accumulate in various organs, causing inflammation and organ damage, and driving Gaucher disease symptoms.
Signs and symptoms
Gaucher disease can affect multiple organs throughout the body, but the spleen, liver, and bone marrow are usually the ones most affected. The most common signs and symptoms of Gaucher disease are therefore related to damage to those organs. They are:
- enlarged spleen (splenomegaly) and liver (hepatomegaly), leading to swelling and tenderness in the abdomen
- low red blood cell counts (anemia), which causes fatigue and weakness
- low platelet counts (thrombocytopenia), leading to easy bruising and difficult to control bleeding
- bone pain, joint problems, skeletal abnormalities, and weakened bones that are prone to breaking.
People with Gaucher disease type 1 may have all the above symptoms, but will not develop neurological problems. By contrast, those with Gaucher disease types 2 or 3 can have the above symptoms, but will also experience neurological problems that can include:
- abnormal eye movements
- delayed development
- seizures
- problems with coordination
- cognitive impairment.
Some rarer Gaucher disease symptoms can also develop as a result of the infiltration of Gaucher cells in organs and tissues. These symptoms may include:
- Gallstones, or hardened deposits of the digestive fluid bile
- metabolic abnormalities
- lung problems
- heart complications
- impaired kidney function
- brown, pigmented skin spots
- enlarged lymph nodes.
People with Gaucher disease are also at increased risk of developing other health conditions, including Parkinson’s disease and certain forms of blood cancer.
Diagnosis
A Gaucher disease diagnosis can be established if enzyme tests confirm the GCase enzyme is reduced and/or genetic tests detect the presence of disease-causing GBA1 mutations. Such tests are usually done on blood or saliva samples.
The main test used to diagnose Gaucher is the beta-glucosidase leukocyte test, which measures GCase enzyme activity in the blood. If activity levels are below 15% of normal, a diagnosis can usually be confirmed.
Genetic tests that look for GBA1 gene mutations may also be used to establish a Gaucher disease diagnosis, or to clinch the diagnosis after enzyme tests.
Other tests, including blood tests, imaging tests, and tests of organ function may help monitor Gaucher disease and establish its severity, but are generally not required to confirm a diagnosis.
Treatment options
The two main types of Gaucher disease treatment are enzyme replacement therapy (ERT) and substrate reduction therapy (SRT). In the U.S., most of these therapies are only approved for Gaucher disease type 1, but one ERT, Cerezyme (imiglucerase), is also indicated for the nonneurological complications of type 3.
ERT works to deliver a working version of the GCase enzyme to patients via routine infusions into the bloodstream. Three ERT products are approved in the U.S. They are:
- Cerezyme is approved for adults and children with types 1 and 3 Gaucher disease
- VPRIV (velaglucerase alfa) is approved for patients of all ages
- Elelyso (taliglucerase alfa) is approved for adults and children ages 4 and older
SRT also helps prevent the toxic accumulation of Gb1, but it does so by reducing the production of the fatty molecule in the first place. Essentially, these oral treatments work much like turning down a faucet when the drain is clogged, helping to prevent the bathtub from overflowing. The two approved SRTs in the U.S. are:
- Cerdelga (eliglustat), which is approved for certain adults with type 1 Gaucher disease
- Zavesca (miglustat), which is indicated for adults with mild to moderate disease for whom ERT is not an option.
Along with ERT and SRT, which address the underlying cause of the disease, people with Gaucher disease often receive supportive care to help address specific disease complications. These may include pain medications, blood transfusions, physical therapy, and orthopedic surgery.
Life expectancy
Receiving early and appropriate treatment for Gaucher disease is essential to improve Gaucher disease life expectancy, which is reduced without treatment. But available therapies are more effective in some Gaucher types than in others, so life expectancy varies substantially based on disease type. In:
- Gaucher disease type 1, where treatments are generally quite effective, people can expect a normal or near-normal life expectancy
- Gaucher disease type 2, where treatments are not effective, children rarely survive beyond age 2
- Gaucher disease type 3, where treatments can help manage non-neurological symptoms, but not neurological ones, life expectancy is reduced, but many patients will live into their 20s or 30s.
Living with Gaucher disease
Living with Gaucher disease can be a complex journey. People with the condition need to stay on top of their treatments, manage symptoms as they arise, and attend regular medical appointments with multiple healthcare providers.
Just like for anyone in the general population, a well-balanced diet and regular physical activity are recommended for people with Gaucher disease. A physical therapist and a nutritionist can help develop exercise and dietary plans that are adapted to a person’s needs and limitations.
Living with a rare disease can sometimes feel isolating, but support is available. Just as important as managing the physical aspects of Gaucher, people should take care of their mental health and seek support when needed.
Organizations such as the National Gaucher Foundation, the Gaucher Community Alliance, and the International Gaucher Alliance offer a range of Gaucher disease resources, including educational materials, financial assistance, and support groups.
Staying connected, informed, and supported can make a great difference in navigating life with Gaucher disease.
Gaucher Disease News is strictly a news and information website about the disease. It does not provide medical advice, diagnosis, or treatment. This content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or other qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read on this website.