

Gaucher disease type 1: What to know
Last updated July 16, 2025, by Marisa Wexler, MS
 Fact-checked by Inês Martins, PhD
Fact-checked by Inês Martins, PhD
Type 1 Gaucher disease is the most common form of Gaucher disease, a rare inherited disorder caused by mutations in the GBA1 gene. Accounting for about 90% of all cases, it is the only Gaucher disease type that does not affect the brain or spinal cord, meaning it does not cause neurological symptoms.
Early diagnosis and ongoing care are key to managing symptoms and improving long-term outcomes. A variety of treatments are available, and with proper management, patients can have a type 1 Gaucher disease life expectancy close to that of the general population.
Signs and symptoms
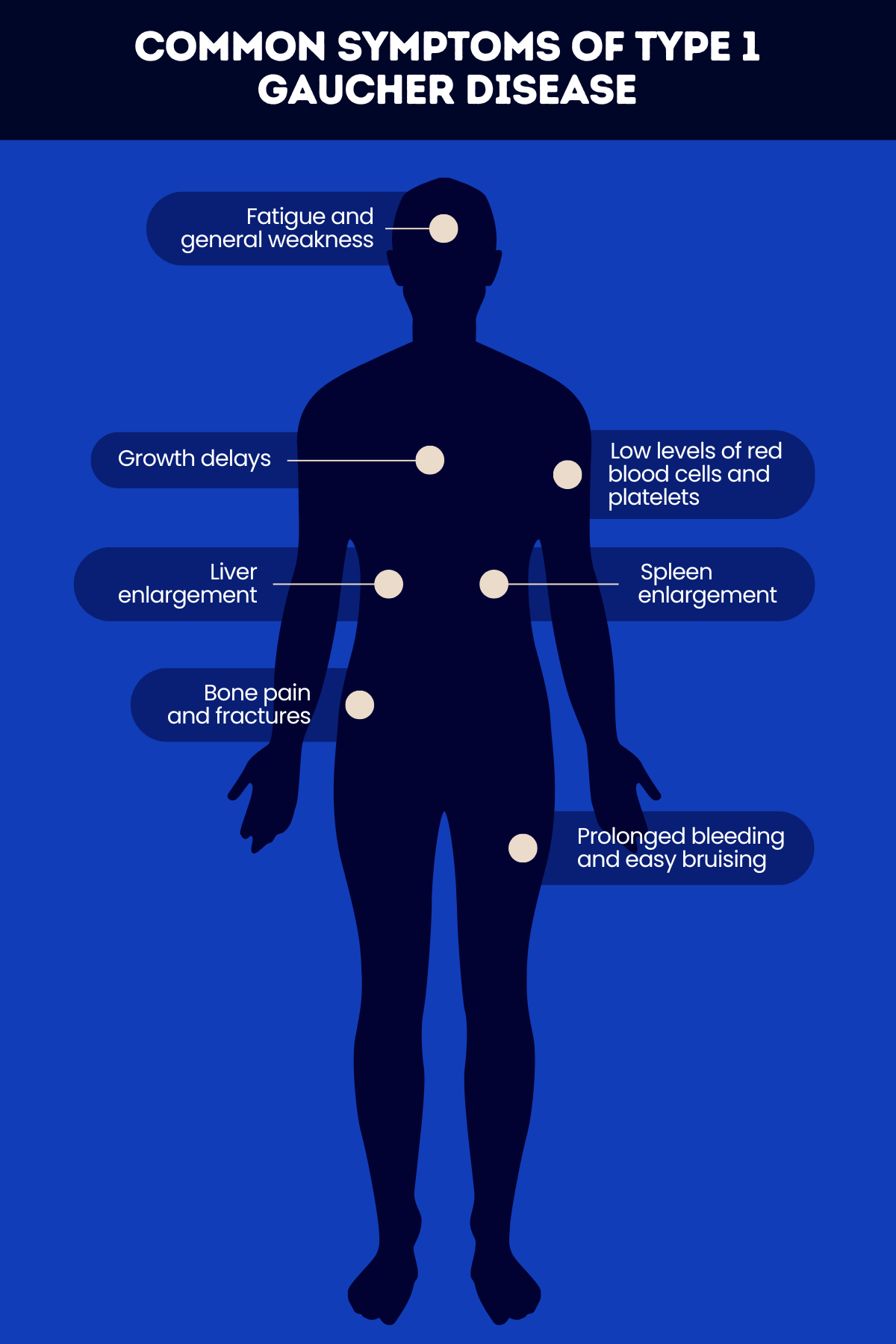
The most common signs and symptoms of type 1 Gaucher disease include:
- liver and spleen enlargement
- low levels of red blood cells and platelets
- fatigue and general weakness
- prolonged bleeding and easy bruising
- bone symptoms, such as bone pain and fractures
- growth delays and delayed puberty.
Type 1 Gaucher symptoms can vary widely from person to person, and may appear at any age — about two-thirds of patients first show signs of Gaucher disease in childhood or adolescence, but others won’t develop symptoms until adulthood.
In general, people who develop symptoms later in life tend to have a milder disease course. There are also some people with Gaucher who never develop symptoms at all.
While Gaucher disease type 1 does not affect the nervous system, patients are at increased risk of developing Parkinson’s disease, a neurological disorder marked by movement problems. Some cancers are also more common in these patients.
Who gets type 1 Gaucher?
Type 1 Gaucher disease affects about 1.5 out of every 100,000 live births worldwide. Because it accounts for the vast majority of all Gaucher cases, this figure closely reflects the overall global prevalence of Gaucher disease.
All types of Gaucher are caused by mutations in the GBA1 gene, with more than 400 mutations having been identified as Gaucher disease causes. However, there is no clear correlation between a person’s specific GBA1 gene mutations and the type of Gaucher disease they may develop. Even identical twins with the exact same genetics can experience very different disease manifestations.
Still, some specific mutations are known to commonly act as type 1 Gaucher causes. For example, people with at least one copy of the p.Asn409Ser or p.Arg535His mutations almost always develop type 1 Gaucher disease.
Gaucher is said to have an autosomal recessive inheritance pattern, which means a person must inherit two faulty copies of the GBA1 gene — one from each biological parent — to develop the condition.
People with only one mutated copy are called carriers. They will not develop the disease but can pass the mutated gene on to their biological children. If two carriers have a child, there is a 25% chance that their child will inherit both mutations and develop Gaucher disease.
Globally, about 0.7% to 0.8% of the population are Gaucher disease carriers. But in some groups, this rate is much higher. For example, around 6% of the Ashkenazi Jewish population, who descend from Jews of Eastern and Central Europe, are estimated to be carriers.
Genetic counseling and carrier screening can help individuals in higher-risk populations better understand their chances of developing or passing down Gaucher disease.
Diagnosis
The process of diagnosing type 1 Gaucher disease is similar to that used for other types of Gaucher. It typically starts with a blood test that measures the activity of glucocerebrosidase (GCase), the enzyme coded by the GBA1 gene. When enzyme activity levels are below 15% of normal, a diagnosis of Gaucher disease is confirmed.
In some countries and U.S. states, Gaucher is included in newborn screening programs. This helps to rapidly identify babies who may develop the disease so they can be referred for further testing and for specialist care if a diagnosis is made.
While enzyme testing can confirm that a person has Gaucher, it cannot distinguish between types 1, 2, and 3. Genetic testing can also identify mutations in the GBA1 gene and support a diagnosis, but it also cannot reliably predict which type a person has.
To make a type 1 Gaucher diagnosis, doctors rely on a careful evaluation of symptoms. If a person has Gaucher disease, but no signs of neurological involvement, type 1 Gaucher disease is usually confirmed.
Treatment options
The two main types of medications used for type 1 Gaucher treatment are enzyme replacement therapy (ERT) and substrate reduction therapy (SRT).
ERT for Gaucher works by delivering a working version of the missing or deficient GCase enzyme through regular infusions into the bloodstream. Three ERT products are approved in the U.S. to treat Gaucher type 1:
SRT for Gaucher works by decreasing the production of fatty substances that accumulate in cells due to GCase deficiency. These therapies are taken by mouth and include:
In addition to ERT and SRT, which can help slow disease progression and improve overall well-being, other treatments may be used to manage certain symptoms or complications of type 1 Gaucher disease:
- pain medications
- blood transfusions to manage low blood cell counts and bleeding
- physical therapy to improve mobility and strength
- orthopedic surgery for joint or bone damage.
Because living with Gaucher disease is different for everyone, treatment plans should be tailored to each person’s needs and preferences.
Living with type 1 Gaucher disease
Gaucher disease type 1 is a lifelong condition that requires regular medical monitoring and personalized treatment. Patients will usually receive care from a multidisciplinary team of specialists, who will work together to address the various aspects of the disease and adjust treatment as needed.
In addition to medical care, healthy lifestyle habits and emotional support play a role in maintaining well-being. These may include:
- eating a balanced and nutritious diet
- exercising regularly
- staying up to date with medical appointments and tests
- seeking mental health support.
Connecting with others who understand the challenges of the disease through Gaucher support groups can also be helpful.
Gaucher Disease News is strictly a news and information website about the disease. It does not provide medical advice, diagnosis, or treatment. This content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or other qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read on this website.



