Types of Gaucher disease
Last updated Jan. 21, 2026, by Marisa Wexler, MS
 Fact-checked by Inês Martins, PhD
Fact-checked by Inês Martins, PhD
Gaucher disease is a rare inherited disorder that’s classified into three main types based on the presence or absence of neurological symptoms, when symptoms first appear, and how quickly they progress.
Regardless of the type of the disease, Gaucher is caused by mutations in the GBA1 gene, which provides instructions for making an enzyme called glucocerebrosidase, or GCase. This enzyme is normally responsible for breaking down certain fatty molecules inside cells.
In Gaucher, low or absent GCase activity causes these fat molecules to toxically accumulate and cause damage to certain tissues and organs. This ultimately drives disease symptoms, but the specific symptoms and their severity can vary substantially from person to person.
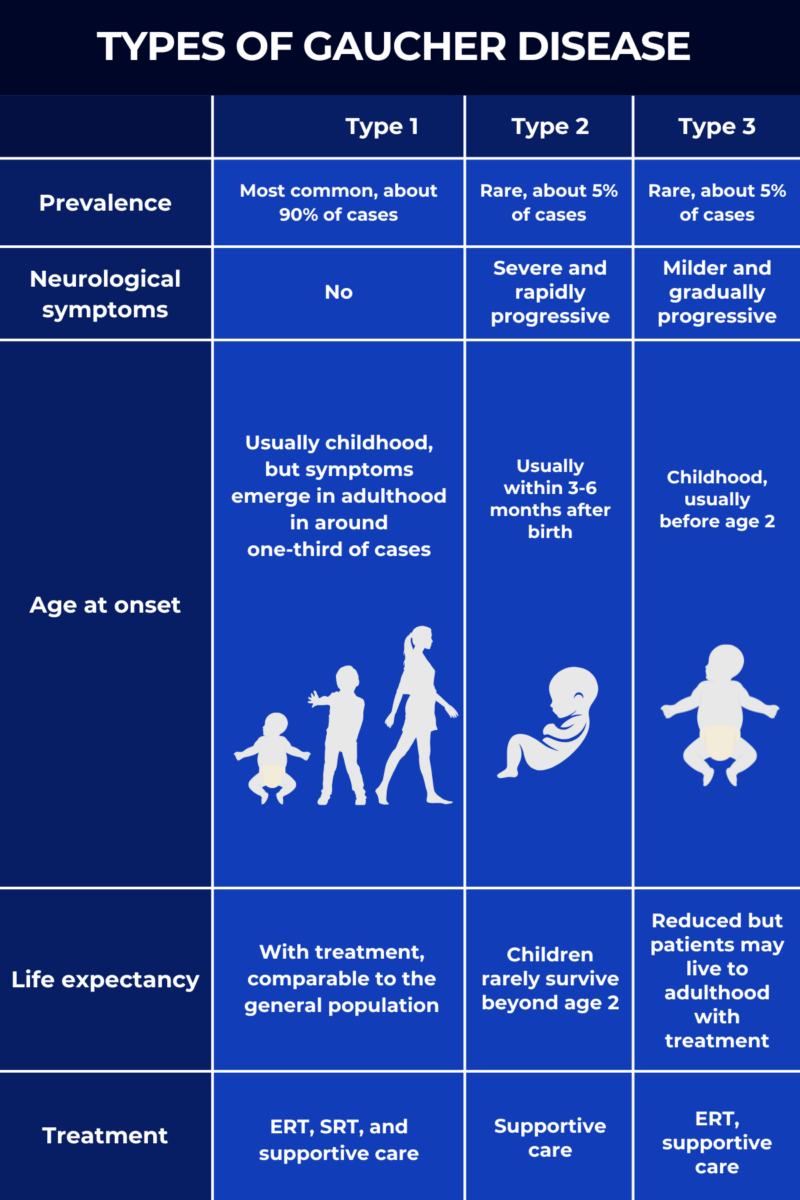
The three types of Gaucher disease are distinguished mainly by the presence and severity of neurological symptoms. Each type also differs in its Gaucher disease life expectancy and response to available treatments.
Different types of Gaucher disease
The three main Gaucher disease types are:
- Gaucher disease type 1
- Gaucher disease type 2
- Gaucher disease type 3.
Each of these types may be further divided into different subtypes. For example, type 2 includes the perinatal-lethal subtype, a particularly severe disease form that affects fetuses or newborns, whereas type 3 can be subdivided into types 3a, 3b, and 3c based on specific disease manifestations.
It’s important to note that Gaucher disease exists along a spectrum of symptoms, which can range from mild or asymptomatic to severe neurological problems. The three Gaucher disease types serve as broad categories to help describe patterns of symptoms, but their divisions may have blurry edges, and some individuals may not fit neatly into one type.
Despite their differences, all types of Gaucher disease will typically cause enlargement of the spleen and liver, as well as blood-related problems such as low blood cell counts and bleeding. The key distinction between the three types lies in their neurological involvement:
- Gaucher disease type 1 is marked by no neurological symptoms.
- Gaucher disease type 2 is marked by severe, early-onset neurological symptoms.
- Gaucher disease type 3 is characterized by milder and gradually progressive neurological symptoms.
While more than 400 mutations in the GBA1 gene have been linked to Gaucher disease, a person’s specific mutations or GCase activity level cannot reliably predict the disease type. Even identical twins who have the exact same genetics can have different manifestations of the disease. For that reason, a Gaucher disease type diagnosis is primarily done by assessing whether neurological problems are present and how severe they are.
Available treatments for Gaucher disease, including enzyme replacement therapy (ERT) and substrate reduction therapy (SRT), can help manage many symptoms of Gaucher disease. However, these therapies cannot reach the brain and spinal cord, so they cannot treat the neurological complications of Gaucher.
Thus, people with type 1 Gaucher disease often experience the greatest benefit from available treatments. Those with type 3 also may see improvements in certain bodywide symptoms, but neurological complications will persist. There are, however, no treatments to slow disease progression or manage symptoms in people with type 2 Gaucher disease.
Type 1 Gaucher disease
Type 1 Gaucher disease is the most common form of the condition, accounting for about 90% of all cases. It is often referred to as non-neuronopathic Gaucher disease because, unlike types 2 and 3, it does not affect the nervous system.
The most common Gaucher disease type 1 symptoms include:
- enlargement of the spleen and liver
- low blood counts, namely low red blood cell and platelet levels
- fatigue and general weakness
- easy bruising and prolonged bleeding
- bone problems such as bone pain and fractures.
Most people with Gaucher disease type 1 — about two-thirds — begin to experience symptoms in childhood or adolescence, and about half are diagnosed before age 10. But approximately one-third of patients don’t develop noticeable symptoms until adulthood. As a general rule, patients who develop symptoms later on tend to have a milder disease course.
The two main Gaucher disease type 1 treatment options are:
- ERT, which delivers a functional copy of the GCase enzyme to help degrade the fatty molecules that accumulate in cells
- SRT, which lowers the production of the fatty molecules that GCase normally degrades, thereby limiting their accumulation.
In addition to these treatments, which target the disease’s underlying cause, patients may benefit from supportive care. This may involve blood transfusions, surgery for bone problems, and pain-relieving medications, each of which may help address specific symptoms and complications.
While type 1 Gaucher disease is considered the mildest form, it can still lead to serious complications, including life-threatening bleeding, liver and bone marrow failure, and an increased risk of certain cancers.
Without treatment, Gaucher disease type 1 life expectancy is reduced by several years. But with appropriate treatment, especially if started before serious complications arise, people with this disease type have a life expectancy that is close to that of the general population.
Gaucher disease type 1 prevalence is estimated at about 1.5 per 100,000 live births worldwide. Because the vast majority of Gaucher cases are type 1, this figure is similar to the overall Gaucher disease prevalence.
Type 2 Gaucher disease
Type 2 is the most severe and rapidly progressive form of Gaucher disease, accounting for about 5% of all cases. Also known as acute neuronopathic Gaucher disease, this type is estimated to affect 2 of every 1 million live births worldwide. However, because most patients do not survive early childhood, the actual prevalence in the global population is close to zero.
Some common Gaucher disease type 2 symptoms, such as spleen and liver enlargement and low red blood cell and platelet counts, are similar to those observed in other disease forms. But type 2 is primarily marked by severe and rapidly worsening neurological symptoms. Among them are:
- abnormal eye movements
- muscle rigidity, especially in the neck, trunk, and legs, which cause the head and legs to arch backward
- difficulty swallowing
- developmental delays and poor growth
- seizures
- trouble breathing.
These symptoms typically emerge within the first 3-6 months of life.
Because there is no approved Gaucher disease type 2 treatment to slow or stop disease progression, supportive care is the only available option, aiming to keep patients as comfortable as possible. Most children with type 2 Gaucher disease do not survive beyond age 2.
Perinatal lethal Gaucher disease
A particularly severe subtype of type 2 is perinatal-lethal Gaucher disease. In this form, babies are either stillborn or die shortly after birth. This is the rarest form of Gaucher disease, and is marked by symptoms such as:
- severe swelling and organ damage caused by fluid buildup in tissues
- enlarged spleen and liver
- thickened, scaly skin resembling fish scales
- multiple joint contractures, or damage to connective tissues, that limit movement
- abnormal facial features or malformations
- dangerously low platelet levels.
There is no approved treatment for perinatal-lethal Gaucher disease. In rare cases, ERT may help slow organ damage and extend survival to a few years, but survival beyond the first few months after birth remains extremely rare.
Type 3 Gaucher disease
Gaucher disease type 3, also called chronic neuronopathic Gaucher disease, is another rare form of the disorder, accounting for about 5% of cases. The estimated Gaucher disease type 3 prevalence is about 1 of every 1 million live births worldwide.
Gaucher disease type 3 symptoms include many of the same ones seen in type 1 disease, such as:
- enlarged liver and spleen
- blood-related issues (e.g., low blood cell counts, easy bruising, bleeding)
- bone pain and skeletal abnormalities
However, type 3 also involves neurological symptoms, which are generally milder and progress more slowly than in type 2. These can include:
- abnormal eye movements
- seizures
- difficulty feeding and poor weight gain
- stiffness and poor coordination
- cognitive impairment and dementia
- trouble breathing
The symptoms of type 3 usually appear later than in type 2. About half of patients experience neurological symptoms in the first or second years of life, but others won’t start to notice symptoms until later on. There have been cases of symptoms developing in adulthood, but these are less common.
It’s also possible that patients are first diagnosed with type 1 Gaucher and then develop neurological symptoms consistent with type 3 disease several years later.
Type 3 Gaucher disease may be divided into three subtypes based on particular patterns of symptoms, though there is substantial overlap between these different subtypes.
- Gaucher disease type 3a is associated with seizures (myoclonic epilepsy).
- Gaucher disease type 3b is marked by milder neurological problems but more severe organ involvement and skeletal abnormalities.
- Gaucher disease type 3c, also called cardiovascular Gaucher disease, is marked by severe heart complications, such as valve and blood vessel calcification, scarring and enlargement of heart muscles, and heart failure.
In the U.S., an ERT called Cerezyme (imuglucerase) is approved as a Gaucher disease type 3 treatment option, and other ERTs can also be used off-label for this indication. This approach helps to ease nonneurological symptoms and improve quality of life for patients, but it cannot stop neurological symptoms from progressing.
Supportive care, such as orthopedic or cardiac surgery and treatments to ease pain, also can help manage specific disease complications.
Because available treatment cannot address the neurological aspects of the disease, Gaucher disease type 3 life expectancy is reduced relative to the general population. Still, with appropriate care, many patients live into their 20s or 30s. Some with milder disease may reach their 40s or 50s.
Comparing the three main types of Gaucher disease
Although all Gaucher disease types are caused by the same genetic defects, the condition can present very differently depending on the specific type. The three main types are distinguished based on the presence and severity of neurological symptoms, but other factors such as prevalence, age at symptom onset, response to treatment, and overall prognosis will also differ.
- Prevalence: type 1 Gaucher disease is by far the most common form, accounting for at least 90% of all cases. Types 2 and 3 are much rarer, each representing about 5% of cases.
- Neurological symptoms: the key distinction between the three types is their presence and severity. Type 1 has no neurological symptoms, whereas type 2 is marked by severe, rapidly progressing neurological symptoms, and type 3 by milder neurological symptoms that progress more slowly.
- Age at symptom onset: in type 2, symptoms typically appear within the first months after birth. In type 3, symptoms usually emerge within the first years of life, and almost always in childhood. By contrast, type 1 symptoms often manifest in childhood, but about one-third of patients still don’t experience symptoms until adulthood.
- Response to treatment: while available therapies for Gaucher are generally effective for managing non-neurological disease manifestations, they cannot address neurological symptoms. As a result, treatments are most effective for type 1. People with type 3 may still benefit from treatment for non-neurological issues, but any treatments will have a minimal effect on those with Gaucher type 2.
- Life expectancy: with appropriate treatment, people with type 1 can expect a normal or near-normal life expectancy. In type 3, treatment may extend life into adulthood, but life expectancy is still substantially reduced. Children with type 2 Gaucher disease rarely survive beyond early childhood.
Gaucher Disease News is strictly a news and information website about the disease. It does not provide medical advice, diagnosis, or treatment. This content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or other qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read on this website.