

Gaucher disease type 3: What to know
Last updated Jan. 21, 2026, by Marisa Wexler, MS
 Fact-checked by Inês Martins, PhD
Fact-checked by Inês Martins, PhD
Type 3 Gaucher disease is a rare form of Gaucher disease that accounts for about 5% of all cases. Also known as chronic neuronopathic Gaucher disease, this type is defined by neurological symptoms that appear in childhood and progress gradually over time.
While there are treatments to help manage non-neurological symptoms in type 3 Gaucher, no therapies are available to slow or stop the progression of neurological complications. As a result, life expectancy is reduced compared with the general population — but with early diagnosis, appropriate care, and regular monitoring, many patients can live into adulthood.
What causes Gaucher disease type 3?
Gaucher disease is an inherited lysosomal storage disorder caused by mutations in the GBA1 gene. This gene provides instructions for making glucocerebrosidase, an enzyme that is needed to break down certain fatty molecules inside cells.
When the GBA1 gene is mutated, it leads to glucocerebrosidase deficiency. As a result, fatty substances accumulate to toxic levels in various organs and tissues, especially the liver, spleen, and bones.
In type 3 Gaucher, this buildup also affects the brain and spinal cord, causing neurological symptoms.
Gaucher disease is inherited in an autosomal recessive manner, which means the disease only develops if a person inherits two faulty copies of the gene — one from each biological parent.
More than 400 GBA1 gene mutations have been linked to Gaucher. While it’s difficult to predict a person’s exact type based on their mutations alone, some mutations are more frequently seen in certain types. One example is the p.Leu483Pro mutation, which is often linked to neuronopathic Gaucher (types 2 and 3), though it has also been reported in type 1.
How is it different from type 1 and type 2?
The key distinction between the three main Gaucher disease types lies in their neurological involvement.
- Type 1, the most common form, is defined by a complete absence of neurological symptoms.
- Type 2 and type 3 are both considered neuronopathic Gaucher disease forms, meaning they affect the brain and spinal cord.
What sets type 3 Gaucher disease apart from type 2 is that its neurological symptoms usually emerge later in childhood, and sometimes even in adolescence or adulthood, and tend to progress more slowly.
This slower disease course allows most people with type 3 to live into adulthood, while those with type 2 generally do not survive beyond early childhood.
While type 3 Gaucher disease prognosis is generally better than for type 2, it is still less favorable than for type 1, where life expectancy is close to that of the general population.
Signs and symptoms
Type 3 Gaucher disease causes symptoms that affect both the nervous system and other parts of the body. These symptoms tend to appear in childhood, with about half of patients experiencing neurological symptoms before age 2, but their onset and severity can vary widely between individuals.
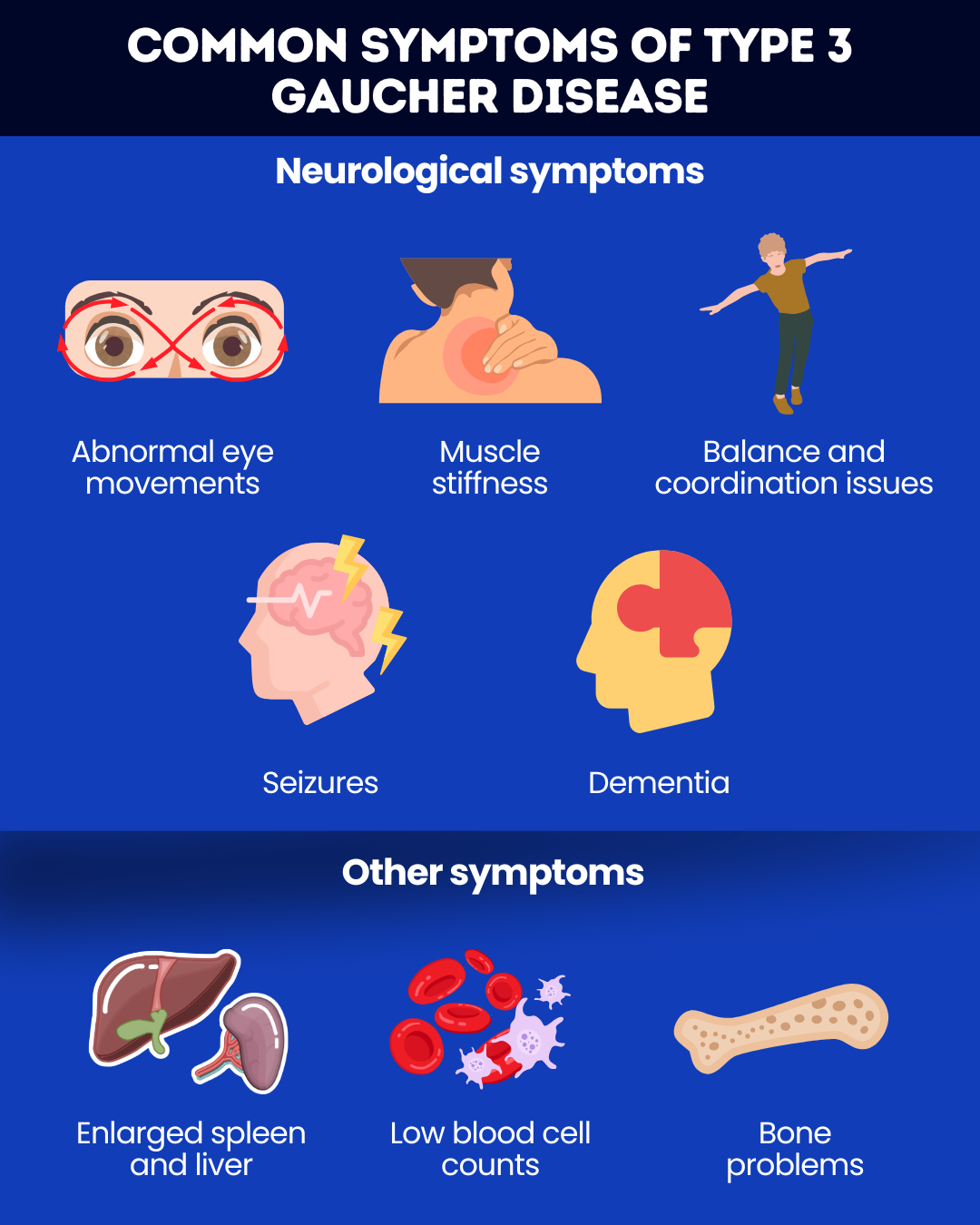
Neurological symptoms of type 3 Gaucher may include:
- oculomotor apraxia, or trouble moving the eyes voluntarily
- muscle stiffness and spasms
- problems with balance and coordination
- seizures
- dementia.
Other common type 3 Gaucher disease symptoms include:
- enlarged spleen and liver
- low blood cell counts, leading to fatigue and easy bruising and bleeding
- bone problems, such as pain and frequent fractures.
While neurological symptoms are the defining feature of type 3, the exact pattern of symptoms can differ. Some children may experience only mild eye movement issues, while others develop more serious neurological complications like seizures or mobility challenges.
How is type 3 Gaucher diagnosed?
A diagnosis of Gaucher disease is usually made by measuring the activity of the glucocerebrosidase enzyme in a blood sample. If activity levels are found to be below 15% of normal, a Gaucher diagnosis can be confirmed.
Genetic testing can also be used to look for GBA1 mutations that cause Gaucher disease and help support the diagnosis. But neither enzyme testing nor genetic testing can determine which type of Gaucher disease a person has.
To diagnose type 3 Gaucher, doctors need to look at how the disease is presenting, particularly whether neurological symptoms are present, and how they are progressing.
If children diagnosed with Gaucher disease show slowly progressing neurological symptoms, doctors may suspect and confirm a Gaucher type 3 diagnosis. Imaging tests and detailed neurological evaluations can help assess the presence and severity of neurological involvement in Gaucher disease children.
Treatment options
While there are no type 3 Gaucher disease treatment options that can slow or stop the progression of neurological complications, several therapies are available to help manage other symptoms and improve quality of life.
Enzyme replacement therapy (ERT), which delivers a functional version of the glucocerebrosidase enzyme to the body, is generally recommended for all people with type 3 Gaucher. Although ERT cannot cross into the brain and spinal cord and therefore does not address neurological symptoms, it can be used to manage non-neurological symptoms such as enlarged organs, blood-related complications, and bone problems.
In the U.S. and some other countries, only the ERT medication Cerezyme (imiglucerase) is formally approved for type 3 Gaucher, but other ERTs may be prescribed off-label.
Another class of Gaucher treatment, called substrate reduction therapy, also may sometimes be used to treat non-neurological symptoms in patients with type 3, but its benefits are generally more limited.
In addition to ERT, care often includes supportive therapies such as:
- pain medications
- blood transfusions
- physical therapy
- orthopedic surgery.
Outlook and life expectancy
Type 3 Gaucher disease prognosis generally falls between that of the other two main disease types. But predicting the exact type 3 Gaucher life expectancy is difficult, because the disease exists on a wide spectrum.
Outcomes vary depending on a number of factors, including the severity of symptoms and the quality of care that patients receive. Still, with appropriate care, many patients live into their 20s or 30s, and some with milder forms of the disease may reach their 50s.
Because the disease is progressive and affects multiple organs, lifelong medical care is essential to manage the disease’s complications. A multidisciplinary care team can help monitor symptoms, manage complications, and provide support at every stage of the disease.
People living with rare genetic diseases like Gaucher type 3 may also benefit from mental health support and patient support groups, which can help them navigate the challenges of a chronic condition and feel less alone.
Gaucher Disease News is strictly a news and information website about the disease. It does not provide medical advice, diagnosis, or treatment. This content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or other qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read on this website.



