

Gaucher disease treatment
Last updated Jan. 21, 2026, by Marisa Wexler, MS
 Fact-checked by Inês Martins, PhD
Fact-checked by Inês Martins, PhD
Treatment for Gaucher disease usually includes therapies that address the underlying cause of the disease, as well as supportive treatments to manage symptoms.
Gaucher disease is a rare genetic disorder caused by mutations in the GBA1 gene. This gene contains instructions to make an enzyme called glucocerebrosidase, known as GCase, which is normally needed to break down a fatty molecule called glucocerebroside, or Gb1.
In Gaucher disease, reduced GCase activity causes Gb1 and related molecules to build up to toxic levels inside certain cells. This ultimately leads to a range of Gaucher disease symptoms, including enlarged organs, blood-related problems such as low blood cell counts and bleeding, and bone abnormalities.
Many of these symptoms are shared across the three main Gaucher disease types, but types 2 and 3 are also associated with neurological symptoms. Gaucher disease type 1 is considered a milder disease form that does not affect the brain and spinal cord, so patients typically do not experience neurological complications.
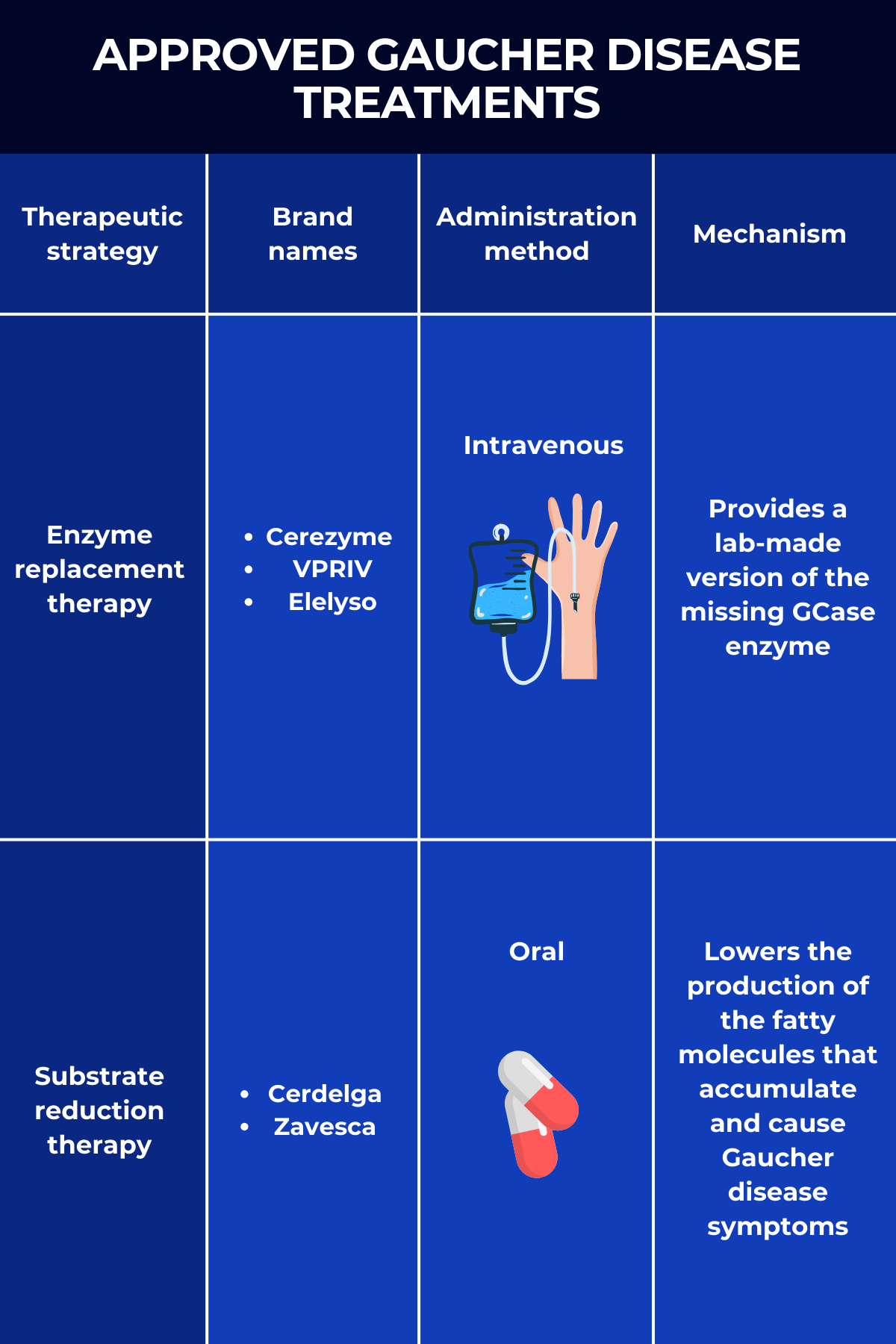
The two main Gaucher disease treatment approaches are enzyme replacement therapy (ERT) and substrate reduction therapy (SRT), both of which work to reduce the toxic buildup of Gb1 in the body.
These treatments are effective for managing many aspects of the disease, but they offer the most benefit for people with Gaucher type 1. Because ERT and SRT cannot enter the brain or spinal cord, they are less effective for treating neurological symptoms associated with types 2 and 3.
Research has shown that starting treatment soon after a Gaucher disease diagnosis is linked to better long-term outcomes, especially for people with type 1 disease. Early treatment also can help prevent disease complications, improve quality of life, and extend survival.
Enzyme replacement therapy
Known simply as ERT, enzyme replacement therapy was the first type of Gaucher disease treatment to gain widespread approval. As the name suggests, it works by providing a functional version of the GCase enzyme that people with Gaucher disease lack.
This replacement enzyme is delivered intravenously, or into the bloodstream. Once there, it is picked up by affected cells — mainly immune cells called macrophages — where it helps break down the toxic Gb1 that drives disease symptoms.
Three ERTs are approved in the U.S. for treating Gaucher disease:
- Cerezyme (imiglucerase), marketed by Sanofi, is approved to treat the non-neurological symptoms of Gaucher disease types 1 and 3 in adults and children. It is the only Gaucher disease therapy approved in the U.S. for people with type 3.
- VPRIV (velaglucerase alfa), by Takeda Pharmaceuticals, is approved for the long-term treatment of children and adults with type 1 Gaucher disease. The enzyme it delivers has the same sequence as the human GCase but has been modified to boost its entry into macrophages.
- Elelyso (taliglucerase alfa), sold by Pfizer, is approved to treat Gaucher disease type 1 in adults and children ages 4 and older. The lab-made enzyme is produced using plant cells, making it inherently efficient at entering macrophages without requiring additional modifications.
Cost
Studies suggest that the average yearly ERT costs range from about $140,000 and $325,000 per patient, depending on disease severity.
Still, because ERT treatments are dosed based on body weight, the overall price of treatment can vary significantly from patient to patient, depending on weight and selected dose. Actual costs to patients also will vary based on insurance coverage and local pricing regulations.
Substrate reduction therapy
SRT, fully known as substrate reduction therapy, is a class of oral treatments that also reduce levels of Gb1 inside cells. It works, however, by preventing the production of the fatty molecule. This is achieved by blocking the activity of glucosylceramide synthase, the enzyme that makes Gb1.
By reducing the production of Gb1, SRT helps prevent its toxic buildup, which in turn alleviates symptoms associated with the disease.
While SRT is generally more convenient and less invasive, it is available only for adults and is typically only used as an alternative to ERT when that approach is deemed unsuitable. It may be used as a first-line therapy for some patients.
There are two approved SRT products for Gaucher disease treatment:
- Cerdelga (eliglustat), sold by Sanofi, is approved for the long-term treatment of adults with Gaucher disease type 1 who are extensive, intermediate, or poor CYP2D6 metabolizers, but not ultra-rapid metabolizers. CYP2D6 is the enzyme that metabolizes Cerdelga. This SRT is the only medication that can be used as first-line oral therapy.
- Zavesca (miglustat), marketed by Johnson & Johnson, is approved to treat adults with mild-to-moderate type 1 Gaucher disease for whom ERT is not an option.
While not approved specifically for Gaucher disease type 3, Cerdelga and Zavesca can be sometimes used to manage non-neurological complications in these patients. Still, because they cannot cross the blood-brain barrier to effectively reach the brain and spinal cord, they do not have a significant impact on neurological symptoms.
Venglustat, a newer SRT by Sanofi that can cross the blood-brain barrier, is under investigation for treatment of neurologic manifestations of this disease type.
Cost
Much like ERTs, the annual cost of SRT for treating Gaucher disease varies based on the specific medication prescribed, as well as a number of other individual factors. These include dosage, insurance coverage, and regional pricing.
There isn’t much data on the costs for Zavesca, but the annual expense for Cerdelga can be as much as $500,000 for intermediate and extensive CYP2D6 metabolizers, who require higher doses for the same effect, and $250,000 for poor metabolizers.
Experimental and emerging therapies
While ERT and SRT are the only approved treatments that target the underlying cause of Gaucher disease, a number of experimental and emerging therapies are being explored.
These novel approaches, some of which may improve treatment outcomes in people with Gaucher disease types 2 and 3 — who are often harder to treat — include:
- chaperone therapy
- gene therapy
- newer SRT versions
- a stem cell transplant.
Chaperone therapy
For the GCase enzyme to break down Gb1 effectively, it must fold into the correct three-dimensional structure. However, some Gaucher-causing mutations result in an abnormally folded enzyme that cannot function properly.
Chaperone therapy aims to correct this misfolding by using small molecules that bind to the misfolded enzyme and try to help achieve the correct structure. In theory, this strategy could offer an alternative treatment for Gaucher disease, though a notable limitation is that chaperone therapy is expected to be effective only in patients with specific types of mutations.
One compound that has been investigated as a potential chaperone therapy for Gaucher disease is ambroxol hydrochloride, a commonly used cough and cold medicine. Early clinical trials have indicated that this treatment may be used to ease Gaucher symptoms in some patients.
Ambroxol hydrochloride is notably able to cross into the brain, so it may be used off-label to help manage neurological complications of Gaucher disease.
Gene therapy
Gene therapy for Gaucher disease involves delivering a healthy copy of the GBA1 gene to a patient’s cells, enabling the production of a functional GCase enzyme.
This approach could, in theory, provide a one-time treatment with long-term benefits, offering a potential cure for the underlying cause of the disease.
Gene therapies for Gaucher disease are still in the early stages of development, but several candidates are being tested in clinical trials. Among them are:
- PR001, which is being developed by Prevail Therapeutics for Gaucher disease type 1
- FLT201, being developed by Spur Therapeutics for Gaucher disease type 1
- VGN-R08b, which Shanghai Tianze Yuntai Biopharmaceutical is developing to treat Gaucher disease types 2 and 3
- LY-M001, which Lingyi Biotech is developing for Gaucher disease type 1 and 3.
Newer SRT versions
Another promising investigational therapy for Gaucher disease is venglustat, an oral SRT being tested in a Phase 3 clinical trial (NCT05222906).
Venglustat is being evaluated in combination with an ERT to treat both neurological and non-neurological complications of Gaucher disease type 3.
Stem cell transplant
Hematopoietic stem cells (HSCs) are stem cells that live in the bone marrow and produce all types of blood cells. Stem cell transplantation — also called bone marrow transplant or, more formally, allogeneic hematopoietic stem cell transplantation — involves collecting HSCs from a healthy donor and transplanting them into someone with Gaucher disease to replace the patient’s dysfunctional stem cells.
In theory, once the donor stem cells make their way into the patient’s bone marrow, they can produce new blood cells carrying a functional version of the GCase enzyme. This means that the newly formed macrophages, immune cells particularly affected by Gb1 accumulation, will be able to break down the fatty molecules that drive Gaucher disease symptoms.
Some small studies have indicated that stem cell transplant may help improve Gaucher symptoms in certain patients. However, the procedure is highly invasive and carries substantial risks, so it is generally only considered for patients with severe disease manifestations who cannot access or tolerate standard therapies. The procedure is also sometimes used for people with severe type 3 Gaucher disease.
Supportive care
Beyond treatments to address the disease’s underlying cause, people with Gaucher disease often receive supportive care to help address disease complications. Supportive care may include:
- pain medications such as non-steroidal anti-inflammatory drugs (NSAIDs) or opioids
- medications and supplements to help promote bone health, such as bone-strengthening agents, calcium, and vitamin D
- drugs to help normalize blood clotting
- blood transfusions to manage low blood cell counts and bleeding
- orthopedic surgery to address bone and joint damage
- physiotherapy to strengthen the body and improve mobility.
A splenectomy, or surgical removal of the spleen, also is a supportive care option, though it is relatively uncommon now. The procedure, however, may help address significant spleen damage or persistently low blood cell counts.
How treatment varies by Gaucher disease type
Gaucher disease types are mainly defined based on the presence and severity of neurological symptoms.
- Gaucher disease type 1, the most common form, is marked by an absence of neurological symptoms.
- Gaucher disease type 2 is marked by severe neurological complications that appear soon after birth and progress rapidly.
- Gaucher disease type 3 is characterized by milder neurological problems that develop in childhood and progress more slowly.
Because available treatments are most effective for addressing non-neurological complications of the disease, how Gaucher disease is treated varies based on a person’s disease type:
- Gaucher disease type 1 treatment usually involves standard ERT or SRT therapies, alongside supportive care to help with symptom management.
- Gaucher disease type 2 treatment essentially consists of supportive care and palliative treatment to make patients and their families comfortable. There are no effective options for managing symptoms or slowing disease progression in these patients.
- Gaucher disease type 3 treatment may involve ERT to manage non-neurological complications of the disease. Supportive care and off-label therapies remain the only option to manage neurological problems.
Research is ongoing to look for new ways to treat the different types of Gaucher disease.
Lifestyle changes that may support treatment
While lifestyle changes cannot address the root cause of Gaucher disease, they can play a meaningful role in maintaining overall health, supporting symptom management, and improving quality of life.
Eating a balanced, nutritious diet can help support energy levels, bone health, and immune function — all of which may be affected by Gaucher disease. Staying physically active, within one’s limits, can also support bone, joint, and cardiovascular health, along with mood. Patients should speak with their healthcare provider to develop a safe and effective exercise plan that suits their needs.
Avoiding tobacco and limiting alcohol consumption is also important for minimizing the risk of complications and supporting the liver and bones, which are commonly affected in Gaucher disease.
It’s also essential that Gaucher patients meet regularly with healthcare providers for routine check-ups. Mental health support — such as talk therapy, support groups, or counseling — can also be beneficial for managing the emotional challenges that can come with a chronic rare disease.
People affected by Gaucher disease may also benefit from connecting with advocacy organizations such as the National Gaucher Foundation, the Gaucher Community Alliance, and the International Gaucher Alliance. These groups offer valuable support and resources, and in some cases, assistance with navigating the cost of Gaucher disease treatment.
Gaucher Disease News is strictly a news and information website about the disease. It does not provide medical advice, diagnosis, or treatment. This content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or other qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read on this website.
Related content
-
-
 Discussion
Discussion
-
-

-

-
Discussion