New Method of Diagnosing and Monitoring Gaucher Disease Uses Dried Blood Samples
Written by |
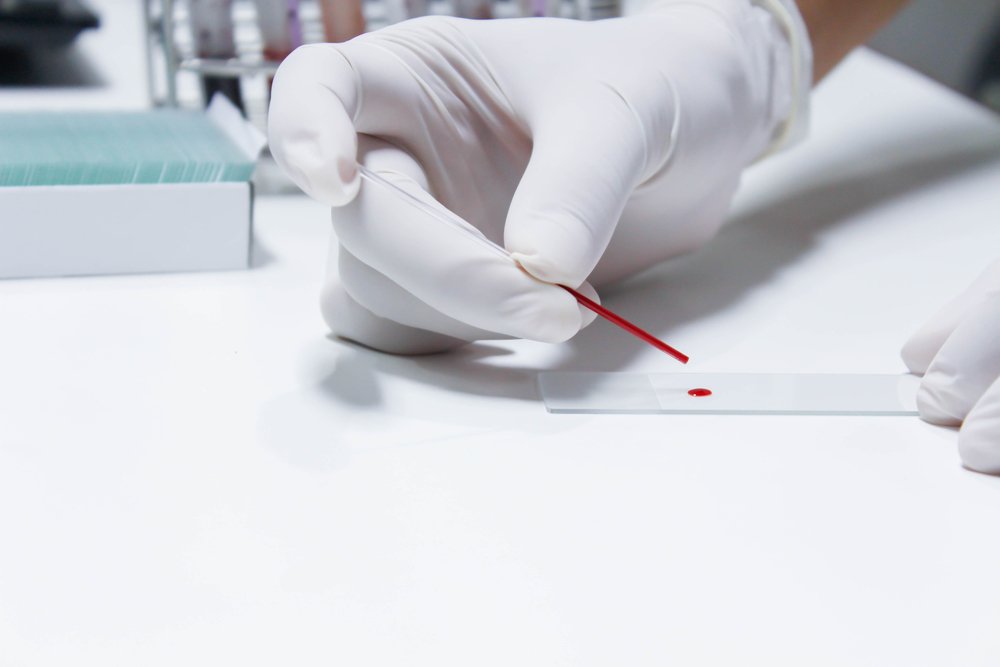
Using dried blood samples for measuring the levels of Gaucher disease biomarker glucosylsphingosine is now possible with a newly developed method by a team of researchers at Cincinnati Children’s Hospital Medical Center.
The study, “A convenient approach to facilitate monitoring Gaucher disease progression and therapeutic response,” appeared in the journal Analyst.
Mutations in the GBA1 gene lead to deficient production of an enzyme called acid beta-glucosidase. This causes the buildup of lipids (fat), particularly glucosylceramide and glucosylsphingosine, in several organs and tissues including the lungs, liver, spleen, brain tissues, and in blood circulation.
High levels of circulating glucosylsphingosine previously were suggested as a highly sensitive, reliable, and specific biomarker for Gaucher disease diagnosis. Additionally, its levels were proposed to help monitor disease progression and the effectiveness of therapies. In a prior study, researchers found that glucosylsphingosine levels in a Gaucher disease patient were 100-fold higher when compared with a healthy control.
The authors of this study tested the effectiveness of a new method for detecting glucosylsphingosine directly in dried plasma spots (DPS), essentially a dried sample of blood. Using DPS samples has several advantages, including its low cost and easy processing. The samples also can easily be shipped from distant point-of-care clinics to central laboratories.
The method is based on tandem mass spectrometry, a technique that can accurately identify a molecule, such as a glucosylsphingosine, by measuring each molecule’s intrinsic mass-to-charge ratio.
The new method was then tested in DPS collected from Gaucher disease patients. The researchers analyzed plasma spots from 19 GD-related human samples shipped from Brazil and Thailand. In total, 10 samples were from Gaucher disease type I, four were type III, and one was type II. Additionally, three samples were analyzed from Gaucher disease carriers, who have one defective gene and one normal gene. These carriers do not have the disease but can pass the Gaucher gene to their children.
“All type I GD patients except one pregnant woman were on therapy when specimens were collected,” the authors reported.
The sample analyses showed that in nine patients with Gaucher disease type I who were taking enzyme replacement therapy (ERT), glucosylsphingosine levels were reduced to a mean level of 31 nM, which is “much lower compared to a pre-treated specimen at level of 85.8 nM, but still significantly elevated compared to healthy controls,” they explained. Also, the levels of glucosylsphingosine in three treated patients with the disease type III were also lower relative to an untreated patient.
Researchers tested the method in two different mouse models for Gaucher disease. One model (known as 9V/null mice) is characterized by brain alterations and neurological deficits at late age and progressive glucosylceramide and glucosylsphingosine accumulation in the brain and in visceral tissues. In the other model (referred to as 4L;C*) glucosylceramide and glucosylsphingosine accumulate mainly in the central nervous system.
While showing comparable levels of plasma glucosylsphingosine, the 4L;C* mice had much higher glucosylsphingosine accumulation in the brain than those of 9V/null mice.
Overall, “we describe a robust and convenient tandem mass spectrometry assay for the measurement of the Gaucher disease biomarker, glucosylsphingosine in dried plasma spots,” the authors wrote.
“The utilization of dried plasma spots permits convenient shipment and transport of patient samples between sites thus enabling not only monitoring of disease progression in GD patients to be performed but also assessment of response to therapies,” they added.